

A team of HIV researchers, cellular biologists, and biophysicists who banded together to support COVID-19 science determined the atomic structure of a coronavirus protein thought to help the pathogen evade and dampen response from human immune cells. The structural map – which is now published in the journal PNAS, but has been open-access for the scientific community since August – has laid the groundwork for new antiviral treatments tailored specifically to SARS-CoV-2, and enabled further investigations into how the newly emerged virus ravages the human body.
“Using X-ray crystallography, we built an atomic model of ORF8, and it highlighted two unique regions: one that is only present in SARS-CoV-2 and its immediate bat ancestor, and one that is absent from any other coronavirus,” said lead author James Hurley, a UC Berkeley professor and former faculty scientist at Lawrence Berkeley National Laboratory (Berkeley Lab). “These regions stabilize the protein – which is a secreted protein, not bound to the membrane like the virus’s characteristic spike proteins – and create new intermolecular interfaces. We, and others in the research community, believe these interfaces are involved in reactions that somehow make SARS-CoV-2 more pathogenic than the strains it evolved from.”
Structural biology in the spotlight

Generating protein structure maps is always labor intensive, as scientists have to engineer bacteria that can pump out large quantities of the molecule, manipulate the molecules into a pure crystalline form, and then take many, many X-ray diffraction images of the crystals. These images – produced as X-ray beams bounce off atoms in the crystals and pass through gaps in the lattice, generating a pattern of spots – are combined and analyzed via special software to determine the location of every individual atom. This painstaking process can take years, depending on the complexity of the protein.
For many proteins, the process of building a map is helped along by comparing the unsolved molecule’s structure to other proteins with similar amino acid sequences that have already been mapped, allowing scientists to make informed guesses about how the protein folds into its 3D shape.
But for ORF8, the team had to start from scratch. ORF8’s amino acid sequence is so unlike any other protein that scientists had no reference for its overall shape, and it is the 3D shape of a protein that determines its function.
[How deep learning can help solve protein structures]
Hurley and his UC Berkeley colleagues, experienced in structural analysis of HIV proteins, worked with Marc Allaire, a biophysicist and crystallography expert at the Berkeley Center for Structural Biology, located at Berkeley Lab’s Advanced Light Source (ALS). Together, the team worked in overdrive for six months – Hurley’s lab generated crystal samples and passed them to Allaire, who would use the ALS’s X-ray beamlines to take the diffraction images. It took hundreds of crystals with multiple versions of the protein and thousands of diffraction images analyzed by special computer algorithms to puzzle together ORF8’s structure.
“Coronaviruses mutate differently than viruses like influenza or HIV, which quickly accumulate many little changes through a process called hypermutation. In coronaviruses, big chunks of nucleic acids sometimes move around through recombination,” explained Hurley. When this happens, big, new regions of proteins can appear. Genetic analyses conducted very early in the SARS-CoV-2 pandemic revealed that this new strain had evolved from a coronavirus that infects bats, and that a significant recombination mutation had occurred in the area of the genome that codes for a protein, called ORF7, found in many coronaviruses. The new form of ORF7, named ORF8, quickly gained the attention of virologists and epidemiologists because significant genetic divergence events like the one seen for ORF8 are often the cause of a new strain’s virulence.
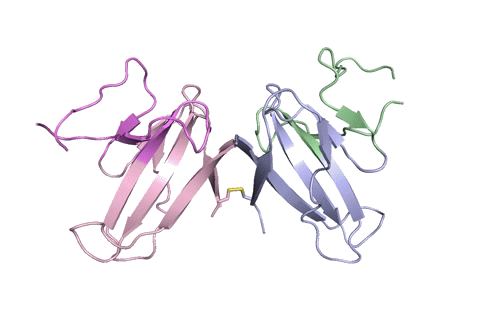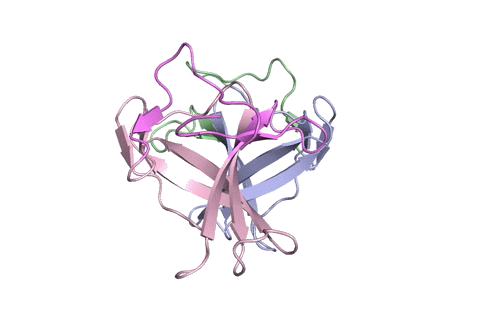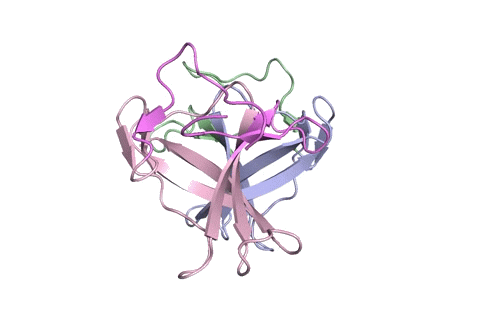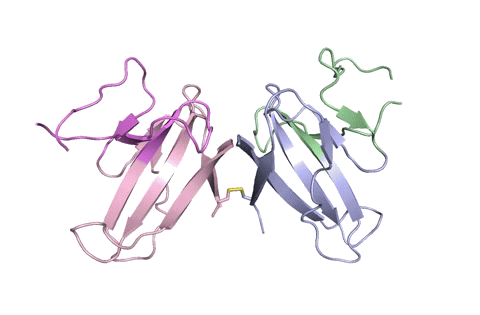
A ribbon diagram of the ORF8 structure. This protein is composed of two units with identical amino acid sequence and shape that are bound together by a sulfur-sulfur bond. (Credit: Hurley Lab)
“Basically, this mutation caused the protein to double in size, and the stuff that doubled was not related to any known fold,” added Hurley. “There’s a core of about half of it that’s related to a known fold type in a solved structure from earlier coronaviruses, but the other half was completely new.”
Answering the call
Like so many scientists working on COVID-19 research, Hurley and his colleagues opted to share their findings before the data could be published in a peer-reviewed journal, allowing others to begin impactful follow-up studies months earlier than the traditional publication process would have allowed. As Allaire explained, the all-hands-on-deck crisis caused by the pandemic shifted everyone in the research community into a pragmatic mindset. Rather than worrying about who accomplished something first, or sticking to the confines of their specific areas of study, scientists shared data early and often, and took on new projects when they had the resources and expertise needed.
In this case, Hurley’s UC Berkeley co-authors had the viral protein and crystallography expertise, and Allaire, a longtime collaborator, was right up the hill, also with crystallography expertise and, critically, a beamline that was still operational. The ALS had received special funding from the CARES Act to remain operational for COVID-19 investigations. The team knew from reviewing the SARS-CoV-2 genomic analysis posted in January that ORF8 was an important piece of the (then much hazier) pandemic puzzle, so they set to work.
The authors have since all moved on to other projects, satisfied that they laid the groundwork for other groups to study ORF8 in more detail. (Currently, there are several investigations underway focused on how ORF8 interacts with cell receptors and how it interacts with antibodies, as infected individuals appear to produce antibodies that bind to ORF8 in addition to antibodies specific to the virus’s surface proteins.)
“When we started this, other projects had been put on hold, and we had this unique opportunity to hunker down and solve an urgent problem,” said Allaire, who is part of Berkeley Lab’s Molecular Biophysics and Integrated Bioimaging Division. “We worked very closely, with a lot of back and forth, until we got it right. It really has been one of the best collaborations of my career.”
From sequence to structure

A ribbon diagram rendering of the ORF8 structure predicted by AlphaFold 2 (blue), overlaid onto the actual structure (green) determined by the UC Berkeley-led team. (Credit: DeepMind)
Sequencing a gene or a string of amino acids to understand the components of a protein is fast and easy for scientists these days, but studying how a sequence of amino acids interact to fold into the protein’s actual physical form using X-ray crystallography or cryo-electron microscopy is complex and time intensive. As a consequence, there has been a longstanding call within biology to develop tools that accurately predict a protein’s structure based on its sequence.
In the past few decades, machine learning has emerged as the front-runner in this challenge. These artificial intelligence programs are fed large datasets of known protein structures so that they learn to identify correlations between sequence and fold shape, quickly finding patterns that would take years for humans to discover. Once the program – called an algorithm – is “trained” in this way, it can be used to build predictive models of unsolved protein structures. And every time it is fed a new confirmed structure, it improves.
To test which algorithms are the best, companies and institutions hold competitions, the most famous of which is the biannual Critical Assessment of protein Structure Prediction (CASP) experiment. Last year, ORF8 was selected as the final challenge of the CASP competition because it “stood out as exceptionally hard to predict,” according to Hurley. The top algorithms were set loose on the ORF8 structure, as well as other structures, and it wasn’t until these structures were released in the Protein Databank in August that the CASP judges were able to select a winner. AlphaFold 2, an algorithm developed by Google offshoot DeepMind, came out on top after constructing structures that most closely matched the experimental targets, including that of ORF8.
The Advanced Light Source is a Department of Energy Office of Science user facility. The Berkeley Center for Structural Biology is supported in part by the Howard Hughes Medical Institute and the National Institutes of Health.
# # #
Founded in 1931 on the belief that the biggest scientific challenges are best addressed by teams, Lawrence Berkeley National Laboratory and its scientists have been recognized with 14 Nobel Prizes. Today, Berkeley Lab researchers develop sustainable energy and environmental solutions, create useful new materials, advance the frontiers of computing, and probe the mysteries of life, matter, and the universe. Scientists from around the world rely on the Lab’s facilities for their own discovery science. Berkeley Lab is a multiprogram national laboratory, managed by the University of California for the U.S. Department of Energy’s Office of Science.
DOE’s Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit energy.gov/science.